24 /10/2014
BỆNH HỆ MÔ BÀO ( HISTIOCYTOSIS)
MỤC TIÊU HỌC TẬP
1.Trình bày được tiêu chuẩn chẩn đoán hội chứng thực bào theo hội nghiên cứu thực bào
2.Mô tả đặc điểm chẩn đoán hội chứng thực bào thể di truyền
3.Mô tả các nguyên nhân thường gặp hội chứng thực bào thứ phát
4.Nguyên tắc điều trị HCTBM
5.Trình bày đặc điểm bệnh Langerhans Cell Histiocytosis (LCH)
NỘI DUNG
1.Đại cương về hội chứng Histiocytosis ( Mô bào)
-Đặc điểm:năm 1987 thành lập hội Histiocytosis , gồm các chuyên gia lâm sàng,bệnh học,miễn dịch học.
-Phân loại bệnh Histiocytosis dựa hình dạng tế bào, dấu ấn bề mặt tế bào, đặc tính hoá học tế bào.Có ba nhóm bệnh thuộc về Hội chứng bệnh mô bào ( Histiocytosis)
- Bệnh liên quan tới tế bào dendritic (dendritic cell –related )
-Langerhans cell- histiocytosis
-Xanthogranuloma
2.Bệnh liên quan tới đại thực bào (macrophage –related )
-Hemophagocytic lymphohistiocytosis:genetic và sporadic
-Sinus histiocytosis with massive lymphadenopathy
3.Bệnh lý mô bào ác tính liên quan monocyte :monocytic leukemia
Bảng 1 .Phân loại của hội chứng Histiocytosis
| Phân loại | Dendritic cell-related | Macrophage-related | Malignant disorder |
| Loại tế bào | Langerhans | Mononuclear phagocyte | Tế bào ác tính của hệ monocyte-macrophage |
| Chức năng tế bào | Trình diện kháng nguyên | Thực bào và trình diện khng nguyn | Mất chức năng chuyn biệt |
| Chẩn đoán
tế bào |
Birbeck +CD1 + | Birbeck -CD1 –
Nhuộm esterase + |
Cấu trúc tế bào ác tính,thường thuộc nhóm II |
| Bệnh | Langerhans cell His | Genetic và Sporadic erythrophagocytic lymphohistiocytosis | Malignant histiocytosis
Acute monocytic leukemia. |
- Hội chứng thực bào máu (Hemophagocytic Lymphohistiocytosis (HLH) )
Hemophagocytic lymphohistiocytosis là hội chứng được hội Histiocytosis chấp nhận từ 1987 gồm có hai thể Familial Erythrophagocytic Lymphohistiocytosis ,sporadic (FHL) và Infection –Associated –Hemophagocytis .
2.1.Các thể bệnh HLH
2.1.1. Hội chứng thực bào di truyền (Genetic hemophagocytic lymphohistiocytosis hay familial hemophagocytic lymphohistiocytosis (FHL) hay tiên phát (primary hemophagocytic lymphohistiocytosis )
.2.1.1.1 Đại cương
Bệnh được Farquhar và Claireaux mô tả lần đầu (1952). Mac Mahon mô tả năm 1963.
. Tỉ lệ mắc bệnh ở Thụy điển là 1. 2/10 6 /năm trẻ <15 tuổi,tại Nhật tì lệ l 1/800.000/năm
. Giới nam/nữ :1
Tuổi phát bệnh:trẻ <1 tuổi chiếm > 80%.Tuy nhiên thể này vẫn có thể phát bệnh muộn
2.1.1.3. Các thể Hội chứng thực bào gia đình ( di truyền, hay tiên phát) FHL
2.1.1.3.1 Bệnh FHL có tính gia đình : bệnh di truyền theo nhiễm sắc thể thường kiểu lặn. Thể FHL có xuất hiện bệnh ở các nhân di truyền đồng hợp tư, dị hợp tử kép hay đôi khi bị đột biến. Bệnh xảy ra do đột biến gen tạo ra các phân tử tham gia quá trình phóng thích hạt lysosome nhằm tiêu diệt tế bào bị nhiễm bệnh.Hậu qủa tế bào mất khả năng tạo các chất trong quá trình diệt khuẩn.,
Bệnh FHL I (OMIM267700) :đột biến (9q21.3-22)
Bệnh FHLII (OMIM603553) : do đột biến gen tạo perforgen ở vị trí 10q 21-2,
Bệnh FHL III (OMIM 608898) do đột biến gen hMunc13-4(17q25) tạo hạt tiêu bào.
Bệnh FHL IV(OMIM 603552) do đột biến gen STX -11(6q24).
Bệnh FHLV (OMIM 613101).
Ngoài ra một số đột biến gen có liên kết tới hội chứng thực bào di truyền như: GS2 (OMIM 607624); CHS1 (OMIM 214500); HSP2 (OMIM 608233); XLP1 (OMIM 308240); and XLP2 (OMIM 300635) . Bệnh XLP là bệnh tăng sinh lympho bào có liên quan giới tính(X-link lymphoproliferative disease XLP), xác định bệnh nhân có đột biến trong gen SAP (SlAM-associated protein). Gen này nằm trên nhiễm sắc thể giới tính Xq25, có nhiệm vụ điều hòa protein liên quan tới sự dẫn truyền dấu hiệu trong lympho T và natural killer.
2.1.1.3.2 Thể FHL liên quan tới bệnh suy giảm miễn dịch tiên phát như X-linked lymphoproliferative syndrome, Chédiak-Higashi syndrome. Griscelli syndrome ,..
2.1.2. Hội chứng thực bào thứ phát ( Secondary Hemophagocytic Lymphohistiocytosis)
2.1.2.1Định nghĩa: bệnh nhân có đủ tiêu chuẩn theo bảng chẩn đoán bệnh lý mô bào và có bệnh lý đi kèm.
2.1.2.2. Các tác nhân đi kèm
_Nhiễm trùng (( infection-associated Hemophagocytic syndrome = IAHS)
*Siêu vi: Epstein –Barr virus , Cytomegalovirus , Herpes simplex virus, HIV, Adenovirus,,Dengue virus, ,Parvovirus,Varicella-zoster virus.
*Vi trùng: Tuberculose, Babesia microti,Bruscella abortus,Enteric gram-negative, Hib,Mycoplasma pneumonia,Staphylococcusaureus Streptococcus pneumonia.
*Vi nấm:Candida albican,Cryptococcus neoformans, Histoplasma capsultum
*Mycobacter:Mycobacterium tuberculosis
*Rickettsial :Coxiella brunetii
*Ký sinh trùng:Leishmania donovani
_ Bệnh lý huyết học ác tính: bạch cầu cấp, lymphoma (lymphoma associated hemophagocytic syndrome = LAHS),
_ Bệnh lý tự miễn :Lupus hệ thống, viêm khớp thiếu niên,
_ Linh tinh: bệnh chuyển hóa ….
2.2. Sinh lý bệnh:
2.2.1. Bất thường hoạt động miễn dịch ( cơn bão cytokines ) do các đại thực bào bị hoạt động quá mức, tiết nhiều cytokines, gây tổn thương mô và tổn thương đa cơ quan,còn NK và TCD8 thì mất khả năng phản ứng ngược ức chế đại thực bào. Hậu quả đại thực bào tiết cytokines, và NK, TCD8 tiết ra nhiều interferon gamma. Các cytokines thường gặp có nồng độ cao trong huyết tương là interferon gamma (Ifn-γ); tumor necrosis factor alpha (TNF-α); interleukins IL-6, IL-10, IL-12; và soluble IL-2 receptor (CD25)
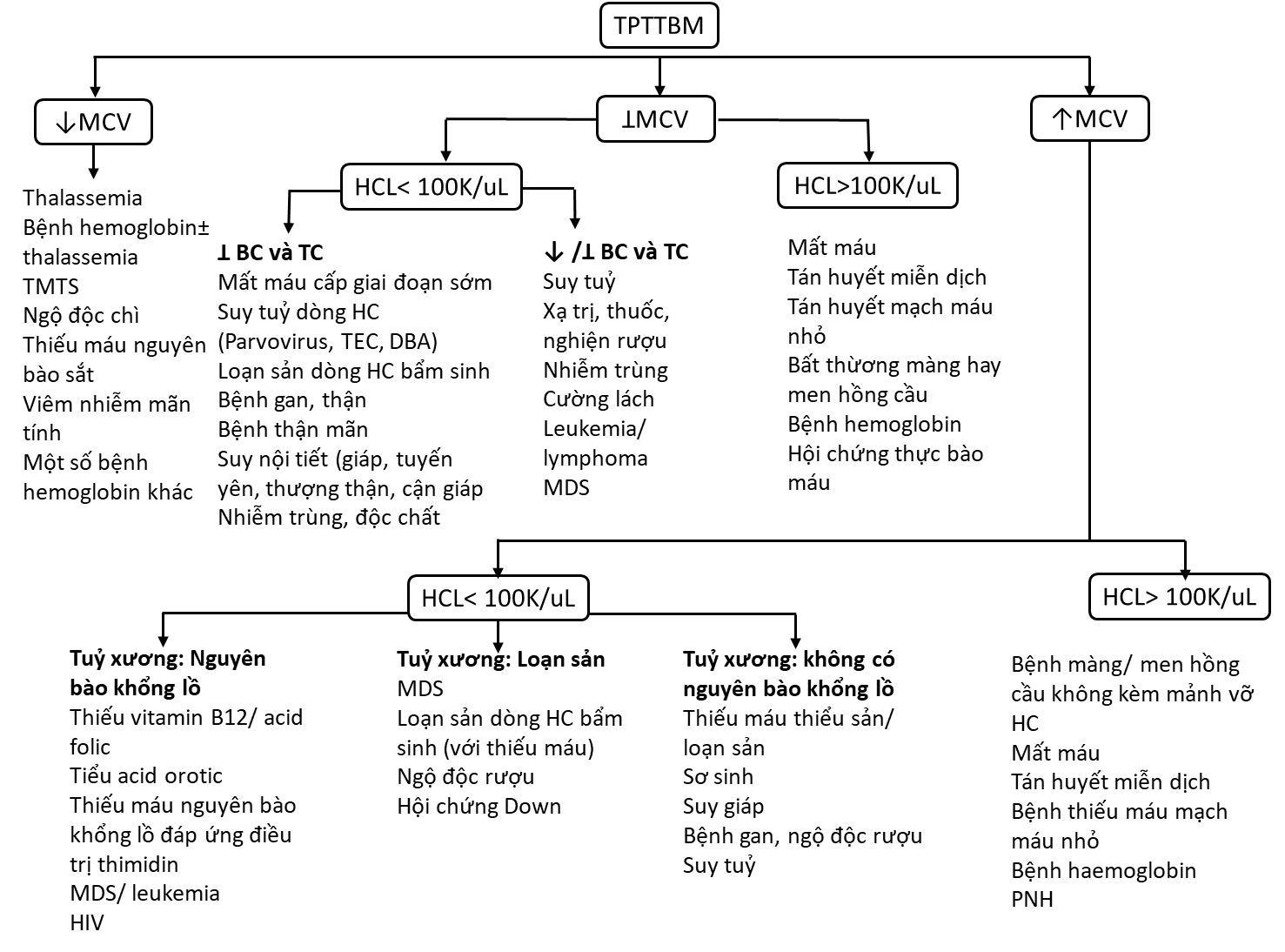
2.2.2. Thực bào máu (Hemophagocytosis): Đại thực bào có vai trò trình diện kháng nguyên và thực bào, cho nên đại thực bào sẽ tăng bắt giữa các tế bào máu ở các vị trí nó hoạt động ( hạch, tủy, gan, lách). Khi có hiện tượng thực bào máu sẽ hỗ trợ chẩn đoán HLH, chứng minh sự tăng hoạt động của đại thực bào.Chỉ có dấu hiệu hemophaocytosis thì không đủ để chẩn đoán
2.3. Triệu chứng lâm sàng và cận lâm sàng
2.3.1.Triệu chứng lâm sàng :Sốt cao liên tục và kéo dài, gan lách to và không đáp ứng kháng sinh điều trị là dấu hiệu gợi ý.Các dấu hiệu thần kinh như li bì, vật vả ,co giật, liệt thần kinh mặt có thể xuất hiện ngay ban đầu. Dấu hiệu vàng da hay phát ban cũng thường xuyên xuất hiện. Thiếu máu hay xuất huyết đặc biệt xuất huyết tiêu hóa cũng có thể xuất hiện sớm nhưng mức độ thường không ồ ạt. Ngoài ra có thể có tổn thương ở hạch cổ ,bẹn, phổi, tim…
2.3.2.Triệu chứng cận lâm sàng
. Huyết đồ: giảm bạch cầu, bạch cầu đa nhân giảm thấp.
.Phết máu ngoại biên xuất hiện bạch cầu không điển hình, hay đơn nhân bất thường.
Tủy đồ: hiện tượng thực bào với sự thay đổi số lượng tế bào tùy vào giai đoạn chọc hút sớm hay muộn. Trong giai đoạn sớm số lượng tế bào tủy vẫn còn bình thường, chỉ xuất hiện tế bào T và ít tế bào thực bào đang hoạt động, giai đoạn muộn số lượng tế bào tủy giảm nhiều, số lượng thực bào tăng cao với hiện tượng thực bào rất ít và không có tế bào ác tính đi kèm. Chức năng gan bị thay đổi với tăng transaminase,tăng trigyceride và cholesterol .
X quang có các thay đổi thường gặp như viêm phổi kẽ, phù phổi hay tràn dịch màng phổi, tràn dịch màng bụng, dầy túi mật, thận phì đại.
Xét nghiệm dịch não tủy : thường có lượng tế bào tăng nhẹ và hoặc tăng đạm. Dịch não tủy thay đổi thường gặp ở bệnh nhân có triệu chứng thần kinh và cũng có thể gặp ở bệnh nhân không có triệu chứng thần kinh (40%), Do đó chỉ định chọc dò tủy sống nên thực hiện trước khi bước vào phác đồ điều trị thực bào máu.
Xét nghiệm cộng hưởng từ ( magnetic resonance tomography= MRT) chỉ định trong trường hợp bệnh nhân có triệu chứng thần kinh hay co giật và hoặc có thay đổi dịch no tủy. MRT phát hiện teo não, dấu hiệu khuyết tán bất thường chất trắng, tổn thương cục bộ chất trắng và chất xám, chậm myeline hóa, vôi hóa nhu mô não.
Xét nghiệm tăng cytokines: soluble CD25 tăng, INF, IL-6, IL-8,IL-10,IL-12, IL-18, TNF-, và macrophage inflammatory protein (MIP 1-).
.Xác định bệnh kèm:tùy hoàn cảnh và diễn tiến của từng bệnh nhân
Nguyên nhân vi trùng,siêu vi, ký sinh trùng….như : cấy máu, cấy dịch não tủy, huyết thanh chẩn đoán, PCR khảo sát. Nên tập trung tầm sóat nguyên nhân nhiễm trùng thường có theo hướng sốt kéo dài tại địa phương: lao, sốt rét ,thương hàn, HIV, HBV, EBV, CMV…
Nguyên nhân bệnh miễn dịch: ANA, anti DNA, tự kháng thể,
Nguyên nhân bệnh ác tính : lymphoma,…
2.4.Tiêu chuẩn chẩn đoán Hemophagocytic Lymphohistiocytosis ( HLH –2004).
Chẩn đoán HLH khi có một trong hai điều kiện sau :
1.Chẩn đoán phân tử xác định HLH
2.Tiêu chuẩn chẩn đoán HLH được xác nhận khi bệnh nhân có đủ 5 trong 8 đặc điểm sau:
A.Tiêu chuẩn chẩn đoán đầu tiên ( HLH 1994) :
Tiêu chuẩn lâm sàng :
1.Sốt >38,5 độ C và trên 7 ngày.
2.Lách to
Tiêu chuẩn xét nghiệm:
3.Giảm 2 trong 3 dòng tế bào :Hb < 9g/dL, Bạch cầu đa nhân trung tính giảm (<1.0×109/L), tiểu cầu <100 x109/L .
Và một thay đổi sau
4.Có một trong hai đặc điểm:giảm fibrinogen máu ≤1.5g/L
Hoặc tăng triglyceride máu khi đói >3.0 mmol/L (hay ≥ 265mg/dL)
Tiêu chuẩn giải phẩu bệnh:
- Có hiện tượng thực bào trong tủy hay lách hay trong hạch. Không thấy dấu hiệu tủy tăng sinh hay u ác xâm lấn tủy
B.Tiêu chuẩn chẩn đoán mới:
6.Hoat lực của tế bào diệt tự nhiên (natural killer) bị giảm hay mất .
- Ferritin ≥ 500microgram/L.
8.CD25 hoà tan(IL-2 receptor ) ≥ 2400 U/ml
Thường bệnh nhân không bộc lộ đủ các triệu chứng trong cùng một lúc, mà các triệu chứng xuất hiện dần về sau. Do đo cần lưu ý để một số hỗ trợ chẩn đoán:
.Nếu lúc nhập viện bệnh cảnh chưa đủ chứng cứ (5/8) thì cần tiếp tục theo dõi .Nếu không khảo sát được tủy đồ thì có thể khảo sát hạch ,gan, lách…Hoặc lập lại tủy đồ sau thời gian theo dõi lâm sàng không đáp ứng .
– Các xét nghiệm có ý nghĩa hỗ trợ chẩn đoán như khảo sát dịch não tuỷ:có tăng tế bào monocytes và hoặc tăng protein.Sinh thiết gan có hình ảnh viêm gan .
-Bệnh nhân có các đặc điểm lâm sàng và xét nghiệm sau: triệu chứng não màng não, hạch to, vàng da, phát ban, men gan tăng, giảm đạm máu, giảm natri máu,VLDL ↑, HDL↓.
–Chẩn đoán dựa khảo sát tủy đồ : có hai histiocytes đang thực bào trên mỗi lam thì xác định chẩn đoán, hoặc có nhiều tế bào T và mô bào (histiocyte) tăng sinh với hemophagocytosis.. Tuy nhiên cần phân biệt nguyên nhân khác có thể hiện tượng thực bào máu.
–Chẩn đoán giải phẩu bệnh xác định tế bào histiocyte qua sinh thiết hạch: quần thể tế bào lympho bị giảm, tế bào histioytes xâm lấn. Hiện tượng này có thể gặp ở các cơ quan khác như lách, gan, não hay thymus. Qua khảo sát dấu ấn miễn dịch tế bào, histiocyte có mang dấu ấn :S-100,CD11c,CD14, CD35, CD68, Immnoglobulin(Ig), Fc receptor,IL-2, phản ứng với kháng thể tới lysozyme và với Mac 387.
–Chẩn đoán di truyền đây là tiêu chuẩn duy nhất để xác định bệnh là thể di truyền: phát hiện bệnh nhân FHL có một trong những thay đổi tại gen.
2.5 Diễn tiến và điều trị
Diễn tiến thay đổi từ vài tuần tới vài tháng,thường hay tái phát.
Điều trị :sau đây là phác đồ để điều trị hội chứng thực bào máu thể di truyền, nhưng nếu bệnh cảnh nặng chưa đủ tiêu chuẩn chính chẩn đoán ,không thể phân biệt được thể di truyền và không thể xác định các bệnh nền kèm theo như nhiễm trùng, miễn dịch, ác tính …thì vẫn được khuyến cáo áp dụng cho đợt điều trị tấn công ban đầu.
2.6. Phác đồ điều trị ban đầu:
Chỉ định: tất cả trường hợp hội chứng thực bào nặng lúc nhập viện,không tìm ra bệnh thực thể đi kèm.
.Mục tiêu điều trị: tăng cường hiệu qủa sống cịn v giảm cc biến chứng của bệnh,giúp lui bệnh.
.Công thức thuốc :Etoposide + Dexamethasone ( bậc thang )+ Cyclosporine A
2.7. Điều trị duy trì
.Chỉ định: tất cả bệnh hội chứng thực bào thể gia đình hay các trường hợp hội chứng thực bào nặng, kéo dài, tái phát. Thời gian này nên chuẩn bị tìm biện phương ghép tủy cho bệnh nhân.
.Mục tiêu điều trị: duy trì tình trạng lui bệnh, giữ cho bệnh không tái hoạt động .
.Công thức thuốc :Etoposide + Dexamethasone ( từng đợt) + Cyclosporine A
2.8.Điều trị cũng cố
.Mục tiêu điều trị :không cho bệnh tái lại nếu không có ghép tuỷ được
.Biện pháp: ghép tuỷ, hoặc chọn lựa một trong các biện pháp
.
2.9. Theo dõi độc tính của thuốc
.Etoposide: lưu ý truyền 0,4mg/ml trong 0,9% Nacl hay Dextrose 5% , TTM trong 1-3 giờ. Lưu ý độc tính có thể gặp: ức chế tủy, hạ huyết áp, tổn thương gan, rối loạn tiêu hóa, đau đầu..
.Dexamethasone: độc tính đói ăn, tăng cân ,bo phì, giữ nước, tăng đường máu. Ức chế tủy, loảng xương, hoạitử, loét dạ dày, viêm tụy, ảnh hưởng thần kinh, cao huyết áp, teo cơ…
.Cyclosporin A: Độc thận, cao huyết áp, nôn ói, loạn sản nướu,tăng transminase, phù, nhức đầu, co giật, ….
3 .Langerhans Cell Histiocytosis (LCH)
3 .1.Lịch sử LCH là một hội chứng phức tạp có ba bệnh đã được mô tả từ lâu với các tên khác nhau :
_Bệnh Hand -Schuller- Christian
1893:Hand mô tả bệnh nhân 3 tuổi có gan lách to,nổi hạch ,lộ mắt và nhiều dấu tiêu xương ở sọ mà lúc đầu ông nghĩ là bị lao.Về sau ông nhận ra Christian và Schuller đã mô tả trước bệnh này nên có tên cả ba người
_Bệnh Letterer Siwe1924 Letterer mô tả bệnh nhi 6 tháng tuổi bị sốt, chảy mủ hai tai,hạch toàn thể ,gan lách to và da bị xuất huyết .Tử thiết cơ quan nội tạng thấy tại sang thương có nhiều tế bào histiocyte được bao quanh bởi các tế bào eosinophil.
1933 Siwe cũng mô tả bé 16 tháng tuổi có gan lách to,hạch toàn thể và tiêu xương ở xương mác .Các nơi tổn thương có kết quả giải phẩu bệnh lý giống như Letterer mô tả,nên bệnh này có tên chung của hai người.
_Bệnh Eosinophil granuloma:Từ 1940 được mô tả ở người kớn và trẻ lớn có tổn thương khu trú ở xương.
_ 1953 Lichtenstein nhận ra cả ba bệnh trên đều có cấu trúc giải phẩu bệnh lý giống nhau là sự hiện diện ưu thế của các tế bào đơn nhân, nên gọi là Histiocytosis X (HX) với X .Các tế bào HX đó chính là tế bào Langerhans nên gọi là Histiocytosis tế bào Langerhans.
3.2.Bệnh căn: chưa rõ do nhiễm trùng hay ung thư hay có thể là sự rối loạn về điều hoà miễn dịch đã làm tăng sinh các tế bào Langerhans.
3.3.Giải phẩu bệnh và miễn dịch học : mô bệnh có nhiều tế bào đơn nhân. Các tế bào này có nhân cuộn lại hay có rãnh ở trung tâm, kèm nhiều hạt nhân, nhiều chromatine, sự biến đổi của nhân tương ứng với giai đoạn tiến triển của bệnh. Ngoài ra còn có các tế bào khác như lymphocyt, eosinophile, neutrophile.
Về miễn dịch học:tế bào Langerhans có dấu ấn S-100 ,CD1a,CD4,CD14, CD74,MHC 1,MHC II đây là các tế bào có nguồn gốc từ tế bào dendritic.Về cytokine thì tế bào của LCH chứa các cytokine như: IL-1α, IL-β.IL-4,GM-CSF,TNF- α và IFN- γ.Ngoài ra khảo sát kính hiển vi diện tử có thể phát hiện hạt Birbeck trong tế bào chất của Langerhans.
3.4.Dịch tễ học
_ Tỉ lệ mắc bệnh :2-5 /M /năm.
_Tuổi :mọi lứa tuổi,đỉnh cao 1-4 tuổi.
3.5.Các thể lâm sàng cổ điển ( không còn sử dụng nữa)
_ Thể khu trú (Eosinophil granuloma= focal dis )
*Đặc điểm khu trú.
*Tổn thương dạng tiêu xương ở một hay nhiều vị trí xương .
*Các xương thường bị là xương dài, xương sọ, đốt sống ,xương mu.
_Thể đa tổn thương (Hand -Schuller- Christian= multifocal dis)
*Đặc điểm: đa tổn thương khu trú.
*Trẻ lớn nhiều hơn trẻ nhỏ.
*Tổn thương toàn thể phối hợp tổn thương nội tạng.
*Tam chứng:tiêu xương,tiểu tháo nhạt ,lộ mắt.
_Thể lan toả (Letterer Siwe =disseminate dis)
*Đặc điểm tổn thương lan toả
*Trẻ dưới 2 tuổi.
*Triệu chứng :sốt kéo dài, sụt cân,tổn thương da, gan,lách ,hạch to, thiếu máu, bệnh lý ở phổi.
Tần suất các cơ quan bị tổn thương lúc chẩn đoán
.Tan xương 80%,
.Hạch to ,sang thương da, gan lách to :50-60%
.Viêm tai giữa và sốt :30-40%
.Tiểu tháo nhạt hay tổn thương phổi :20%
Đặc điểm tổn thương cơ quan
*Xương :
Tổn thương ở một xương nhiều hơn đa xương.
Loại xương:xương sọ, xương hàm, xương dẹp, xương dài.
Dấu hiệu đầu tiên:đau và sưng, diễn tiến nhiều tháng. X quang thấy rõ sang thương.
*Da nổi sẩn đỏ, tróc vẩy, tổn thương tới lớp gai.Thường gặp da đầu, nách, sau tai, tay và chân.
*Đầu cổ: Ở tai gây điếc. Ở xương hàm gây rụng răng. Ở xương hàm trên làm huỷ xương và biến dạng mặt.
*Phổi:Tổn thương đơn thuần hay phối hợp.Thường gặp người lớn .XQ :thâm nhiễm lan toả dạng nốt hay lưới. Lưu ý tổn thương phổi khi bệnh nhân ho, thở nhanh ,tím tái.
*Gan: gan to.Chẩn đoán có tổn thương gan: phù, báng bụng,đạm máu giảm <5,5 g/dL ,albumin <2,5g/dL, bilirubin toàn phần >1,5mg/dL. Không có tán huyết.
*Máu: Hb <10g/dL.BC <4000/mm3.PN < 1500/mm3.Reticulocyte >1,5% .TC < 100000/MM3
Nguyên nhân giảm ba dòng: Cường lách; Tuỷ bị xâm lấn bởi histiocyte
*Nội tiết:Tiểu tháo nhạt xuất hiện khi có tổn thương não.
*Thymus:các tế bào histiocyte tẩm nhuận vào các thuỳ của thymus.
3.6.Chẩn đoán:
.Bệnh sử : bao quát diễn tiến của các triệu chứng đặc biệt như: đau nhức, phù, sẩn da, chảy mủ tai 2 bên, biếng ăn, tiêu chảy, sụt cân, chậm lên cân, khó nuốt, tiểu nhiều, khó thở, các thay đổi về thần kinh và hành vi.
.Lâm sàng: nhiệt độ , vàng da, nhợt nhạt, sẩn da, hạch to, chảy mủ tai, sưng nướu, sưng mô mềm, co kéo liên sườn, gan lách to và ascite bụng.
.Xét nghiệm sàng lọc:
- Công thức máu đầy đủ
- Sinh hóa: protein, albumin, bilirubin, ALT, AST. AP, GT,BUN, creatinine, ion đồ, ferritin.
- Đông máu: PT/INR, aPTT, fibrinogen
- Khảo sát áp lực thẩm thấu và tỉ trọng nước tiểu buổi sáng.
- Siêu âm bụng đo kích thước gan, lách
- XQ ngực, khảo sát hệ xương khi nghi ngờ
- Tiêu chuẩn xác định tổn thương cơ quan trong bệnh LCH khi
Cơ quan có nguy cơ cao
| Cơ quan huyết học | Ít nhất 2 dấu hiệu sau:
thiếu máu Hb <10g/dL ( trẻ nhũ nhi <9g/dL) giảm bạch cầu : <4x 10 9/L giảm tiểu cầu: <100x 1099/L |
| Tổn thương lách | Lách >2cm dưới bờ sườn |
| Tổn thương gan | Gan >3cm VÀ HAY protein máu <55g/L, albumin máu < 25g/L, chẩn đoán GPH |
| Tổn thương phổi | HR-CT và chẩn đoán mô học |
- Tổn thương cơ quan khác:
- Tổn thương sọ
- Tổn thương mắt:sụp mắt, lộ mắt, tổn thương xương hố mắt, cánh bướm, hay xương cánh bướm
- Tổn thương tai; chảy mủ tai ngoài, tai giữa,
- Tổn thương vòm họng: tổn thương vùng họng, nướu, hàm trên và hàm dưới.
- Nguy cơ tổn thương não:
.Xét nghiệm chuyên sâu sau khi có kết qủa sàng lọc
- Tùy vị trí tổn thương được phát hiện, ca1cxe1t nghiệm chuyên biệt được sử dụng để hỗ trợ chẩn đoán như:
- Bảng: Chỉ định xét nghiệm chuyên sâu
| Chỉ định | Xét nghiệm chuyên sâu |
| Giảm 1 hay ba dòng ngoại biên kéo dài | Tủy đồ |
| Tổn thương gan | Sinh thiết gan |
| Tổn thương phổi (XQ ngực)
CT ngực bất thường |
CT ngực,
Rửa khí phế nang tìm tế bào CD1a |
| Tổn thương xương sọ, hàm | MRI đầu |
| Tổn thương xương cột sống | MRI cột sống |
| Bất thường thị giác hay thần kinh | MRI đầu,khám thần kinh, khảo sát neuropsychometric assessment |
| Chảy mủ tai, giảm thính lực, tổn thương xương chủm | Khám thính lực
MRI đầu HR-CT (high resolution- CT) xương thái dương |
| Tiêu chảy kéo dài, chậm lớn, kém hấp thu | Nội soi và sinh thiết |
.Chẩn đoán xác định:
-Sinh thiết hạch hay da hay gan có biểu hiện tăng sinh tế bào Langrhans ở mô bệnh ( CD1a và hoặc CD207 dương ).
.Chẩn đoán mức độ lan rộng: huyết đồ, tủy đồ, x ray hệ xương, CT no hay MRI no.
Chẩn đoán phân loại bệnh LCH theo Hiệp hội bệnh mô bào thế giới
Bảng : phân loại bệnh.
| LCH tổn thương một hệ thống
Single system LCH (SS-LCH) |
Có tổn thương ở một cơ quan hay một hệ thống
(uni –or multifocal) -tổn thương xương cục bộ hay nhiều ổ hay nhiều ổ xương -Tổn thương da -Hạch to -Lung -tuyến hypothalamus- tuyến yên/ hệ TKTU -khác |
| LCH tổn thương đa cơ quan
Multisystem LCH (MS-LCH) |
Trên hay 2 cơ quan bị tổn thương |
3.7. Diễn tiến:
Lâm sàng, điều trị và tiên lượng tuỳ vào mức độ lan rộng của bệnh,có tổn thương đa cơ quan và tuổi khi mắc bệnh.
*Thể cục bộ:ít tái phát, sống lâu.
*Thể đa tổn thương: nguy cơ tái phát cao, nếu không có tổn thương nội tạng tiên lượng tốt.
*Thể có tổn thương nội tạng :tử vong cao.
3.8.Điều trị:
-Chỉ định điều trị toàn thể khi:
SS-LCH với CNS –risk “ lesson
SS-LCH với multifocal bone tổn thương
SS-LCH với vị trí đặc biêt
MS- LCH có cơ quan : risk organ
4.Malignant Histiocytosis Syndrome
-1966 Byrne và Rappaport đặt tên cho hội chứng bao gồm bạch huyết cấp dòng monocyte và lymphoma tế bào histiocyte .
-Lâm sàng: xuất hiện đột ngột ,diễn tiến tối cấp với tổn thương nhiều cơ quan cùng lúc: sốt cao, mệt mỏi, hao mòn, gan,lách to, hạch to. Bệnh nhân có thể kèm theo tổn thương da, phổi, màng tim,xương và não.
-Xét nghiệm:
_Máu:HC giảm, tiểu cầu giảm, BC tăng hay giảm ,có mononuclear atypic.
_Gan:bilirubin tăng, phophatase kiềm tăng
-Chẩn đoán:sinh thiết hạch có thấy tế bào histiocyte.
Chẩn đoán bạch huyết cấp dòng monocyte AML type 5 :tủy có >30% tế bào nhuộm esterase + và CD 14